


 13720223167
13720223167



这些抗生素的糖单元上一般有多个羟基和氨基基团,因此具有水溶性好、功能稳定、抗菌谱广以及与其他抗菌药物协同作用等一系列优点,从而作为化学药物广泛用于治疗各种类型细菌的感染,包括革兰阴性菌和阳性菌[3]。
随着这类药物使用量的不断加大和应用频次的不断增加,氨基糖苷类抗生素出现了不可避免的严重耐药性[4],同时由于其普遍存在的不可逆的耳毒性和肾毒性等毒副作用也限制了氨基糖苷类药物的使用[5]。
虽然随着β-内酰胺类、喹诺酮类、头孢类等毒副作用更少的广谱抗生素的出现,使得氨基糖苷类药物在临床上的应用逐渐减少,但是对于具有多重耐药性的病原菌感染,氨基糖苷类抗生素仍然具有良好的治疗效果,特别是与β-内酰胺类抗生素具有良好的协同效应,使其成为临床上抗击病原菌严重感染的利器[6]。
因此围绕氨基糖苷类抗生素开展的一系列研究一直被国内外相关领域学者关注,特别是随着近年来生物科学技术水平的发展,人们对氨基糖苷类抗生素的认识更加深入[7]。
本文就近年来对氨基糖苷类抗生素的耐药机制和结构改造研究进行综述。
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
1
氨基糖苷类药物的抑菌机制
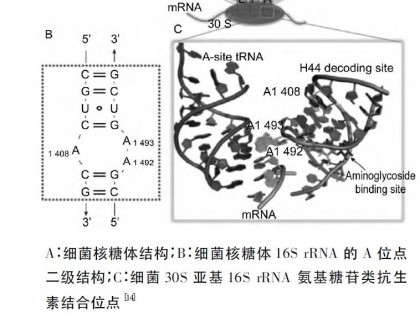
氨基糖苷类药物通过与细菌核糖体30S 亚基的16S rRNA 解码区A 位点特异性结合来发挥作用,使16SrRNA 形成稳定的内环结构[8],如图1 所示。

与此同时,核糖体长期处于活动状态,使得非互补配对的tRNA 也能够通过A 位点,最终导致错误蛋白的形成[9]。
研究表明,除上述作用外,不同的氨基糖苷类化合物由于结构不同,导致其对于A 位点的亲和力也不同,因此在影响核糖体构象的作用上也有着不同的方式,这就造成了活性上的差异[10]。
研究表明,氨基糖苷类药物与A 位点的亲和力对杀菌能力的影响是次要的,而这种结合效应导致的A1492 移动性减弱才是决定其药效更为重要的因素[11]。
除此之外,美国2 个实验室都发现了氨基糖苷类抗生素与核糖体30S 亚基的结合造成的核糖体构象变化会影响细菌核糖体亚基迁移性[12],这种作用抑制了核糖体再循环因子的结合,减慢了核糖体再循环过程,从而影响蛋白质的合成过程[13]。
2
细菌对氨基糖苷类药物的耐药机制
导致细菌产生耐药性的因素很多,包括:
① 细胞外膜通透性改变、内膜转运降低及主动外排系统高表达引起的胞内药物累积浓度降低[15]。
②通过氨基糖苷类抗生素钝化酶( aminoglycoside modifyingenzymes,AME) 对氨基糖苷类抗生素进行修饰[16]。
③ 通过突变或者甲基化修饰改变氨基糖苷类抗生素在细菌核糖体30S 亚基中16S rRNA 上的结合作用位点、作用靶点等。
细菌过度表达氨基糖苷修饰酶是该类抗生素产生耐药性的主要机制[17]。
氨基糖苷类抗生素的氨基和羟基与细菌的核糖体发生一系列相互作用,而它们同时也是病原菌中AME 的作用靶点,如图2 所示。
被修饰后的氨基糖苷类抗生素与细菌核糖体的亲和力减弱,从而产生耐药性[18]。
随着细菌对氨基糖苷类化合物吸收的减少以及各种氨基糖苷修饰酶的出现,氨基糖苷类药物的效果大幅度降低,尤其是氨基糖苷修饰酶在耐药过程中发挥了极大的作用[19]。
修饰酶主要包括氨基糖苷乙酰转移酶( aminoglycoside acetyhransferase,AAC) 、氨基糖苷核苷转移酶( aminoglycosidenucleotidyhransferase,ANT 或AAD) 和氨基糖苷磷酸转移酶( aminoglycoside phosphotransferase,APH) 3大类; 分别对氨基糖苷类化合物上一些特定位置的活性基团进行O-磷酸化、O-腺苷化和N-乙酰化,钝化氨基糖苷类抗生素[20]。
1987 年,Noller 实验室报道了16S rRNA 上A位点的突变及特异性甲基化修饰会造成细菌对氨基糖苷类抗生素高度耐药[21]。
16S rRNA 甲基转移酶通过对A 位点核苷酸进行特异性甲基化修饰,使A位点上的A1408G 发生突变,造成氨基糖苷类抗生素与细菌核糖体结合能力急剧下降[22]。
近年来,研究者在不同病原菌中发现了由质粒介导的16SrRNA 甲基转移酶[22],这种转移酶能特异性甲基化A1408,导致病原菌对氨基糖苷类抗生素高水平耐药。
这一结果说明这种甲基转移酶在各个病原菌中的传播可能是造成近年来氨基糖苷类抗生素出现耐药现象的重要原因之一。
氨基糖苷类化合物抗感染机制和耐药机制的研究成果为设计和开发出抗菌作用更强、毒副作用更少的新型氨基糖苷类抗生素衍生物提供了线索。
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
3
氨基糖苷类药物进展
虽然可以通过寻找全新抗生素来部分解决细菌对抗生素的耐药性问题,但通过对已知氨基糖苷类抗生素进行结构改造来恢复其抗菌活性仍然是一种重要手段。
为了研发出低毒性、抗耐药、高效的氨基糖苷类抗生素,人们不断探究氨基糖苷类化合物抗菌的作用机制,从而根据其抗菌机制,不断对天然氨基糖苷类化合物进行结构改造和创新,尽可能地筛选出既能够紧密结合到细菌核糖体上来干扰细菌蛋白质的合成,达到抗感染效果,又不会被氨基糖苷修饰酶催化改造的新一代的氨基糖苷类化合物。
随着对氨基糖苷类抗生素作用机制与耐药性机制的深入研究,结合核糖体结构与AME 作用机制研究而设计的第二代半合成氨基糖苷类抗生素衍生物的开发也随之进入高潮,如: 基于卡那霉素改造而来的地贝卡星( dibekacin,1971 年) 、阿米卡星( amikacin,1972 年) 、阿贝卡星( arbekacin,1973 年) 。
基于庆大霉素和西索米星( sisomicin) 改造而来的异帕米星( isepamicin,1975 年) 、奈替米星( netilmicin,1976 年) 都已成功投放市场[23]。
到目前为止,研究开发具有耐药菌作用的新的氨基糖苷类抗生素的最有效方法是药物化学方法,即根据构效关系,在已知化学结构上进行各种化学修饰。
虽然氨基糖苷类化合物种类繁多,但结构上都由氨基糖与环醇组成,分为天然的和氨基被修饰的半合成糖苷类化合物,通常以取代的2-DOS 为中心单元,与2 ~ 3 个糖单元通过糖苷键连接而成。
如图3 所示,除链霉素外,新霉素和卡那霉素是最常用的2 种氨基糖苷类抗生素。
从其化学结构可知,新霉素型氨基糖苷类抗生素是以2-脱氧链霉胺( 环Ⅱ)为核心的O-4 和O-5 位糖基化衍生物,而卡那霉素型糖苷类则是以环Ⅱ为核心的O-4 和O-6 位双糖基化的衍生物。
尽管这2 种抗生素结构在环Ⅲ和( 或) R 取代环上有一些微小的差别,但其在rRNA上的结合位点相同[23]。
① 磷酸转移酶修饰。
20 世纪60 年代至70 年代早期,由Umezawa 领导的科研小组开始对磷酸转移酶作用位点进行结构修饰,设法保护卡那霉素免遭APH( 3') 对这一抗生素的钝化作用。由此开发了地贝卡星。但是,APH( 3') -1 酶虽然没有合适的位点,但也能牢固地与3'-脱氧氨基糖苷结合,从而产生耐药性。
② 酰基转移酶修饰ACC( 6') -1。
在酰基转移酶中,6'-N 酰基转移酶的ACC( 6') -1 不仅能修饰天然的卡那霉素和妥布霉素,也同样能修饰N-1 取代的衍生物如阿卡米星和奈替米星等。
③N-1 取代。
用氨酰基或者烷基来取代N-1 氨基的化学修饰。由于天然产物丁酰苷菌素在N-1 含有氨酰基而对很多钝化酶产生抗性。因此开发出具有临床应用价值的阿卡米星、伊帕米星和阿贝卡星。
④ C-1 取代衍生物。
在2-脱氧链霉胺C-1 进行的一系列侧链取代中,庆大霉素的C-1 位的羟甲基取代的产物能够对抗临床上对庆大霉素产生耐药的耐药菌钝化酶的作用,但是同样对类似结构氨基糖苷类抗生素C-1 进行修饰却不能得到同样的结果。
另外可以通过尝试改变手性碳分子结构,例如对西索米星的5-OH 从平伏键改为竖直键,使苷元4-C 和6-C上糖基旋转自由度增大,从而达到对钝化酶产生抗性的作用。
此外,对氨基糖苷类抗生素进行卤代修饰试图避免钝化酶作用,同时卤素的吸电子特性保护临近的基团,但是在避免遭受钝化酶作用的同时,也大大降低了其对敏感菌的活性[24]。
3.1 plazomicin( ZEMDRI)
目前,正在临床前研究的能阻断AME 作用位点及低毒性的第3 代氨基糖苷类抗生素中,最具代表性的是由美国Achaogen 公司开发的plazomicin,如图4 所示。
2015 年,美国FDA 授予plazomicin 合格传染病治疗产品资格,完成Ⅲ期临床试验,并于2018 年6 月25 日获批,以商品名Zemdri 上市。
它以西索米星为前体通过化学合成而来,结合了多种氨基糖苷类抗生素的结构特点以增强其抗菌活性[25]。
在小鼠体外耳蜗外植体的实验清楚地证明了,通过在单一衍生物中联合使用2 种修饰,实验药物plazomicin 能够有效地消除核糖体的选择性、具有与西索米星程度相当的耳毒性以及抗高尔基体自身抗体( anti-Golgi autoantibodies,AGAs) 的活性[26]。
图4 plazomicin 化学结构式
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
3.1.1 plazomicin 药动学研究
注射plazomicin 10 min后血药浓度达到平均峰值,但重复给药时缺乏药物蓄积,推荐治疗剂量为15 mg·kg-1[27]。Plazomicin 的半衰期短( 3 ±0.3) h,清除速率快,为( 1.1 ±0. 1) mL·min-1·kg-1,因此安全性好,不良反应较小,且能快速消除[28]。
Plazomicin 不经肝微粒体或肝细胞代谢,( 89 ± 6) %经尿液排出,在中度和严重肾功能损伤患者中用药时需要调整剂量。
在肾损伤受试者的plazomicin 药动学研究中,血浆浓度在多相物质中下降并且在中度和重度肾功能障碍的受试者中更高[29]。
一项纳入24 例平均年龄为63.9 岁的受试者,在30 min 内接受单次7.5 mg·kg-1静脉内输注plazomicin 的研究表明,肾功能正常的患者为( 37. 9 ±5) mg·L-1,严重肾功能损害者为( 41. 4 ± 7. 83) mg·L-1。
患者根据肌酐清除率( 使用Cockcroft-Gault 公式计算) 分类为肾功能正常[CLcr > 90 mL·min-1],以及轻度、中度或严重损害( CLcr 分别为60 ~ 89,30 ~ 59,15 ~ 29 mL·min-1) 。
另外还发现,对于前两类患者,在24 h 后,plazomicin 浓度低于1 mg·L-1;然而,中度和重度肾功能不全患者的浓度约为10mg·L-1。
轻度肾功能不全受试者和肾功能正常受试者的AUC0 - ∞值相似,其值分别为138 和136 mg·h·L-1; 而中度和重度肾功能不全受试者的AUC0 - ∞值较高,与肾功能正常的受试者相比分别为281 和647 mg·h·L-1。
同样,与肾功能正常的受试者相比,中度肾功能不全( 2. 25 L·h-1) 和严重肾功能不全( 0. 96 L·h-1) 受试者的清除率( CL) 显著降低( 4. 64L·h-1) 。因此,上述药动学结果表明,在中度或重度肾功能不全患者中使用时,需要调整用药剂量[30]。
12 和20 例受试者分别单次静脉注射10. 7 /15mg·kg-1后,在18 ~ 65 岁健康志愿者中进行的一项双盲、随机对照的Ⅰ期临床研究对plazomicin 的肺渗透进行了评估。
通过上皮细胞衬液( epithelial liningfluid,ELF) 的支气管镜微量采样评估肺穿透,估计ELF 与血浆AUC 的比率约为13%,类似于正常受试者支气管分泌物中阿米卡星报告值的范围( 9% ~ 21%) [31]。
此外,plazomicin 在人体中血浆蛋白结合率低( 约12%) ,类似于早期的氨基糖苷类抗生素[32]。
3.1.2 plazomicin 临床试验
一项Ⅱ期多国、双盲、随机、比较对照研究,比较了plazomicin 与左氧氟沙星在复杂性尿路感染( 包括急性肾盂肾炎) 中的作用。
145 例患者被随机分为3 组,分别为每24 h 静脉注射plazomicin 10 或15 mg·kg-1或静脉注射左氧氟沙星750 mg,qd,共5 d,每组分别22,76 和47 例患者。
79 例( 54. 5%) 患者有急性肾盂肾炎,66 例( 45. 5%) 有复杂性尿路感染。
最后一次治疗后( 7 ± 2) d,治愈试验微生物根除和临床的评估结果表明: 针对肠杆菌科分离株的plazomicin MIC 范围为0. 12 ~ 8 μg·mL-1,MIC50和MIC90值分别为1 和2μg·mL-1; 而左氧氟沙星MIC 范围为≤ 0. 12μg·mL-1至> 4 μg·mL-1,MIC50和MIC90值为≤0. 12疗过程中相似,并且发现针对校正意向评估人群( MITT) 的低剂量和高剂量的plazomicin 为67% 和71%,左氧氟沙星组为66%,而各组微生物学疗效评价( ME) 患者的微生物根除率分别为86%,89%和81%。
这些数据提供了plazomicin 在复杂性尿路感染和急性肾盂肾炎中对肠杆菌科的临床疗效的初步证据,包括耐药菌株,因为临床治愈率与早期的氨基糖苷类药物相当[33]。
在一项共有609 例因复杂性尿路感染住院治疗的成人患者( 包括肾盂肾炎) 随机分组的多国双盲试验中,比较了plazomicin( 15 mg·kg-1静脉注射,qd,30 min, iv) 与美罗培南( 每8 h 静脉注射1 g,30 min, iv) 。
在最少4 d 和最多7 d 的静脉注射治疗后,允许改用口服抗菌药物,如左氧氟沙星,共治疗7 ~ 10 d。
在微生物修饰的意向治疗( mMITT) 群体中评估有效性,其包括接受研究药物治疗且有至少1 例尿路病原体感染的患者。
mMITT 人群由388 例患有复杂性尿路感染的患者组成,其中162 例( 41. 8%) 患有肾盂肾炎。
Plazomicin 组和美罗培南组分别在25 例( 13. 1%) 和23 例( 11. 7%) 患者中发现伴随菌血症。
d 5 的复合治愈作为临床复杂性尿路感染消退或改善症状和根除的微生物学结果,见表1。
疗效检测随访时的复合治愈[使用第1 剂研究药物后( 17 ± 2) d]作为临床复杂性尿路感染症状消退和根除的微生物学结果[34]。
plazomicin 组72. 0% ( 18 /25) 的患者和美罗培南组56. 5%( 13 /23) 的患者在伴随菌血症的个体疗效检测随访时达到复合治疗。
plazomicin 组51 /189( 27%) 患者中,有52 种基线肠杆菌科分离株对庆大霉素或妥布霉素或二者都不敏感,如表2 所示,plazomicin 组微生物根除率达到78. 9%( 41 /52) [35]。
3.1.3 plazomicin 的肾毒性
604 例患有尿路系统感染( 包括肾盂肾炎) 的患者随机分组后进行多国双盲非劣效试验,比较plazomicin ( 15 mg·kg-1,30 min iv,qd) 与美罗培南( 1 g·8 h-1,30 min, iv) 。
在iv 治疗4 ~ 7 d 后( M = 5. 1 d) ,可以转换为口服抗菌药,如左氧氟沙星。结果表明,使用plazomicin 的患者中,7% ( 21 /300 ) 血清肌酐增加> 0. 5 mg·mL-1,而美罗培南组为4% ( 12 /297) 。
在静脉注射期间,两组该事件发生率分别为3. 7% ( 11 /300) 和3. 0%( 9 /297) 。
静脉注射完成后的最后一次随访( 8 ~ 43 d) 时,大部分因接受plazomicin 治疗而发生血清肌酐升高的患者完全恢复,美罗培南组则为100%( 9 /9) 。
说明plazomicin 和美罗培南都具有肾毒性且plazomicin 更为严重,由此造成的肾毒性可在之后恢复,美罗培南恢复程度更高。纳入339( 56. 1%) 名女性,年龄中位数为62. 1 岁,99. 3% 为白种人,2% 患者因为不良反应退出试验( 6 /303,6 /301) 。
为保证试验效果,排除了所有CLcr < 30mL·min-1的患者,基线情况无统计学差异。
研究对597 例患有尿路系统感染( 包括肾盂肾炎) 的患者进行随机分组。
年龄、性别及人种无统计学差异,在30 < CLcr≤90 mL·min-1的复杂性尿路感染患者中,9. 7%( 20 /207) 接受plazomicin 治疗的受试者血清肌酐比基线水平升高0. 5 mg·dL-1; 在接受美罗培南的患者中,该比例为4. 1% ( 9 /217) 。
而在CLcr > 90 mL·min-1的患者中,该比例分别为1. 1%( 1 /93) 和3. 8% ( 3 /80) 。
说明在中轻度肾功能损害患者中,plazomicin 的肾毒性比美罗培南更明显,而在重度肾功能损害的患者中则相反。
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
3.2 4'-O-烷基巴龙霉素衍生物
瑞士苏黎世大学医学研究所( Meditinische Mikrobiologie)2015 年设计合成了巴龙霉素的一系列衍生物,并对其进行生物活性研究。
与巴龙霉素相比,引入具有2 或3 个碳的最佳大小的4'-O-烷基,如图5 所示,对野生型细菌核糖体的活性轻微丧失,但选择性显著增加。
巴龙霉素衍生物利用多态性rRNA 残基的额外优势来增加靶选择性。
3.2.1 体内感染实验
第二轮中性白细胞减少免疫抑制后24 h,使用耐甲氧西林金黄色葡萄球菌( MRSA) 临床分离株MRSA AG041 约每只1 × 107CFU 进行小鼠侧耳尾静脉内注射,使小鼠感染。
该菌株MIC 值如下: 巴龙霉素为4. 0 mg·mL-1; 4'-O-乙基巴龙霉素衍生物8 mg·mL-1; 4'-O-丙基巴龙霉素衍生物16 mg·mL-1; 利奈唑胺1. 0 mg·mL-1。
2种巴龙霉素衍生物治疗可有效减少血液和肾脏的细菌负荷,并且在受试最高剂量下,2种巴龙霉素衍生物以及巴龙霉素可将肾脏的细菌负荷降至可检测限度以下。
3.2.2 体内耳毒性
该衍生物的体内耳毒性较庆大霉素明显好转。采用体内氨基糖苷类诱导的听力损失的豚鼠慢性耳毒性模型来比较4'-O-乙基巴龙霉素衍生物和4'-O-丙基巴龙霉素衍生物的耳毒性与庆大霉素的标准耳毒性[36]。
用不同剂量氨基糖苷类处理豚鼠2 周,并通过听觉脑干反应( ABR) 测量12 和32 kHz 的听力阈值来评估耳毒性。
100 mg·kg-1庆大霉素对阈值变化( 即听觉功能丧失) 几乎没有影响,但160 mg·kg - 1 庆大霉素的ABR 阈值超标,而4'-O-乙基巴龙霉素衍生物和4'-O-丙基巴龙霉素衍生物处理对ABR 阈值没有影响,即使在高达400 mg·kg - 1 的剂量下也是如此。
实验还量化了ABR 测量后收获的耳蜗中外毛细胞的损失,在耳蜗表面制剂中看到的毛细胞损失与从ABR 测量获得的功能结果一致。
在用140 mg·kg-1庆大霉素处理的动物中,在耳蜗基部观察到外毛细胞大量损失,与观察到的高频听力损失一致。
相反,在用400 mg·kg-14'-O-乙基巴龙霉素衍生物或4'-O-丙基巴龙霉素衍生物处理的动物中,发现少量( 4'-O-乙基巴龙霉素衍生物) 或没有( 4'-O-丙基巴龙霉素衍生物) 外毛细胞损失。
相比常用的氨基糖苷类庆大霉素、4'-O-乙基、4'-O-丙基巴龙霉素衍生物诱导的听力损失非常小,即使药物浓度过高也是如此,且体内治疗导致很少或没有毛细胞损失。
但是,其肾毒性迹象与庆大霉素相比没有统计学变化[37]。
3.3 TS2037
TS2037 由明治精工制药株式会社的药物研究中心合成,如图5 所示。
TS2037 对抗由抗性金黄色葡萄球菌产生的AAC( 6') -APH( 2″) 氨基糖苷6'-N-乙酰转移酶和2″-O-磷酸转移酶比庆大霉素更稳定。
TS2037 在MRSA 全身感染小鼠模型中的治疗效果优于阿贝卡星、万古霉素和利奈唑胺。
TS2037 对铜绿假单胞菌产生AAC 引起的全身感染的疗效优于阿贝卡星和阿米卡星。
TS2037 在体外和体内针对MRSA 和铜绿假单胞菌实验的氨基糖苷类中表现出最有效的抗菌活性,但是其肾毒性风险仍有待改善。
3.3.1 体外活性
T2037 对MRSA 的杀菌活性实验中,将TS2037 与阿贝卡星、庆大霉素、万古霉素和利奈唑胺对5 株MRSA 的抑制作用进行比较,TS2037( MIC: 0. 5 ~ 1 μg·mL-1) 、阿贝卡星( MIC:1 ~ 4 μg·mL-1) 、庆大霉素( MIC: 64 ~ 128 μg·mL-1) 、万古霉素( MIC: 1 μg·mL-1) 和利奈唑胺( MIC: 2 μg·mL-1) ,TS2037 显示出比阿贝卡星和庆大霉素更强的杀菌活性。
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
3.3.2 耐药性
另外使用液相色谱-质谱法通过电喷雾电离方法检测和测量,从金黄色葡萄球菌RN4220 /pMF490 中的双功能AAC( 6') -APH( 2″) 。
氨基糖苷类修饰酶修饰TS2037、阿贝卡星、庆大霉素和卡那霉素B,结果显示庆大霉素和卡那霉素B被AAC ( 6') -APH ( 2″) 显著修饰。
存在0. 3 mg·mL-1蛋白质时,大多数庆大霉素和卡那霉素B 被修饰为2″-O-磷酸化和6'-N-乙酰化。
另一方面,TS2037 和阿贝卡星抵抗AAC ( 6') -APH( 2″) 的修饰,即使在高浓度( 1. 0 mg·mL-1蛋白质) 时,TS2037也表现出更高的稳定性。
3.3.3 动物实验
鉴定TS2037 的体外和体内活性之间的关系,评估了TS2037 对由MRSA 或铜绿假单胞菌引起的全身性感染小鼠的保护作用。
其中TS2037 对MRSA MF126 和MRSA MF535 的半数有效量( ED50) 分别为2. 65 /0. 85 mg·kg-1,低于阿贝卡星( 17. 8 /21. 0 mg·kg-1) 、庆大霉素( 8. 22 /300mg·kg-1) 、万古霉素( 20. 2 /5. 85 mg·kg-1) 和利奈唑胺( 20. 1 /18. 0 mg·kg-1) 的ED50。
TS2037 对铜绿假单胞菌PAO1 和绿脓杆菌MSC3151 的ED50分别为0. 38 /7. 10 mg·kg-1,是氨基糖苷类抗生素中最低的。
3.3.4 肾毒性
通过测定LLC-PL1 细胞释放NAG( N-乙酰基-β-D-氨基葡萄糖苷酶) 来判断TS2037 和阿贝卡星的肾毒性,在分别用2. 5 和5 μmol·L-1TS2037 处理后,LLC-PK1 的NAG 释放分别为2. 0和2. 1 U·L-1,高于阿贝卡星的1. 1 和1. 3 U·L-1。该结果表明,TS2037 的肾毒性风险高于阿贝卡星[38]。
4
结语
氨基糖苷类抗生素自问世以来一直在临床治疗中发挥重要作用。
虽然其使用因毒性及耐药性受到一定限制,但作为治疗革兰阴性菌导致的严重感染的首选药物在临床中仍然有着不可替代的作用。
随着近年来科学技术的发展,人们对其作用机制、耐药机制有了更加深入的了解,研发出一批新型氨基糖苷类抗生素衍生物,激发了其在临床应用上的新活力。
相关领域的工作者将不断深入了解氨基糖苷类化合物,从分子水平对其作用机制和耐药机制进行具体剖析,用可能出现的新靶点、新机制完善现有资料,为经典药物的改造提供理论支持,从而开发出毒性小甚至无毒副作用的新型氨基糖苷类药物。
同时,药物结构的改造应该积极学习新知识、敢于运用新方法,给老药注入新活力。
参考文献
详见 中国新药杂志2019 年第28 卷第15 期



 加载中...
加载中...